In our last update, I described the rejection of our comparative genomic method paper based on the concerns of a single reviewer. These concerns were clearly addressed both in figures and in the main text. However, we could have made the presentation clearer and more considerate to a broader audience of, e.g. interested (but potentially overworked) non-experts.
Today, I’m happy to report that that our paper has been accepted to Genes, Genomes, and Genetics (G3) under the title “Rock, paper, scissors: harnessing complementarity in ortholog detection methods improves comparative genomic inference.” They say that rejection tends to improve the quality and impact of manuscripts (see write-up). In our case, I think that the extra time and improved focus on clarity and approachability absolutely improved the presentation of the method.
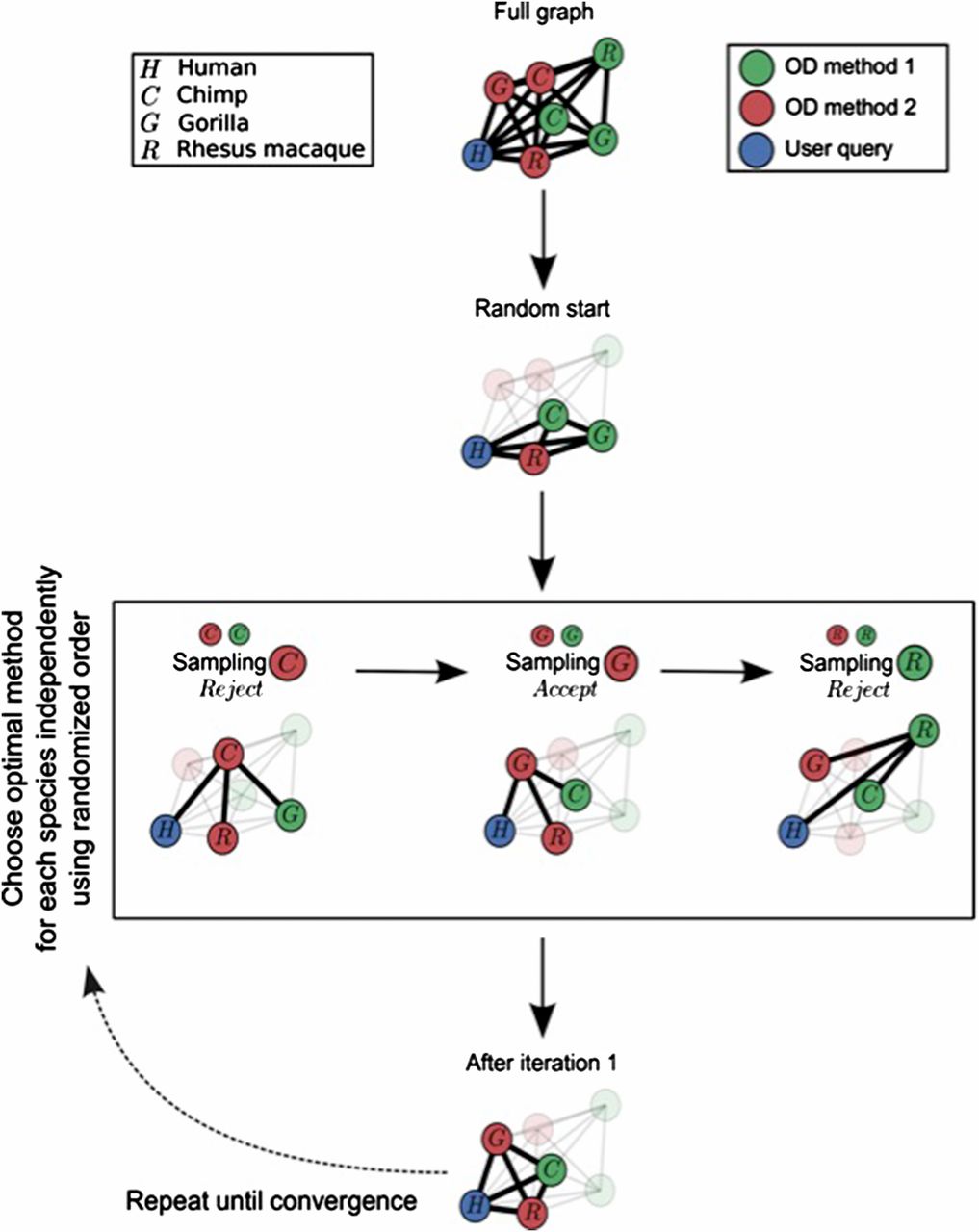
A bit of background on the paper: comparative genomics often relies on the comparison of evolutionarily related sequences from different species (orthologs). Many methods analyze orthologous sequences, but what happens if the orthologs themselves are poorly called? In our experience, this leads to a “garbage in, garbage out” scenario. For this reason, we were motived to improve ortholog detection by building a statistical framework for harnessing the deep complementarity between methodologically diverse ortholog detection methods.
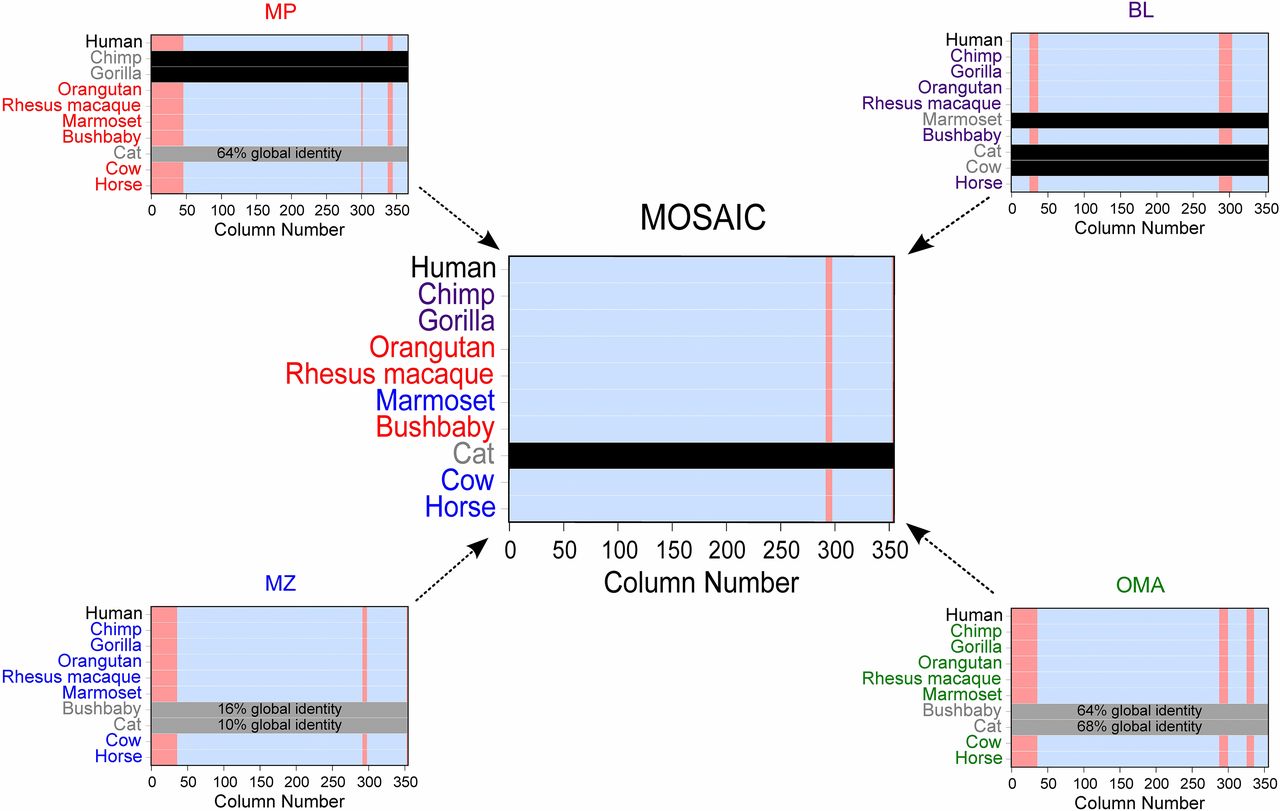
In “Paper, Rock, Scissors”, 1.) we demonstrate this methodological complementarity, 2.) we develop a tool for taking advantage of it, 3.) we show that our tool maintains or improves data quality across wide variety of measures while also retrieving a great deal more sequences, and 4.) apply our method to find an intriguing and otherwise undetectable positively selected site in TPSAB1, an enzyme linked to asthma, heart disease, and irritable bowel disease.